Menu
▾
▴

Home
ChemTraYzer -- Reaction Models from Molecular Dynamics Simulations
ChemTraYzer (CTY) is an integrated simulation and analysis tool to study reactive molecular dynamics of a gas phase.
How to run the ChemTraYzer:
The main script is simulation.py which takes a configuration file as input:
python simulation.py < example/sim.conf
Available settings are desribed in the configuration file (example/sim.conf). CTY then builds the box with packmol and runs the dynamics with LAMMPS. Reactions and species are registered and sent to an quantum mechanics engine for optimization (Gaussian by default).
Once the high level calculations are done, CTY can build the reaction mechanism, and fit Arrhenius and NASA parameters:
python master.py . -reac default.reac -source QM
where default.reac is the reaction list output from simulation.py and QM is the folder with the QM engine output. Final output is the reaction list default.reac, the observed rate constants reac.rate.tab, time-wise species and reactive event counts reac.spec.tab and reac.reac.tab, the mechanism in ChemKin format Default.therm and Default.chem, and the mechanism in a graph format Default.gml.
It is possible to run CTY without the dynamics part, if there is a LAMMPS-style bond table as in example/bonds.tatb:
python processing.py example/bonds.tatb -i 1:6 -i 2:1 -i 3:8 -norec 5
where the repeated -i provides the atom/element mapping and -norec 5 enables the recrossing detection for an interval of 5 data points in the bond table. CTY then creates the reaction list output (bonds.tatb.reac), which in turn can be used to calculate rate constants from the observed events and concentrations:
python analyzing.py examples/bonds.tatb.reac
Since in this case, CTY lacks the geometry information from the dynamics, a QM optimization and thermochemistry parameter fitting is not possible.
How to change the QM engine or the QM submission scripts:
The method submitQM() in simulation.py creates and submits a batch job containing the input for the QM engine (Gaussian09 in our case). It uses some static variables from the Simulation class to fill in e.g. the QM method. If you change the QM engine to something else than Gaussian, you also need to change the QM output read routine harvestGaussian() in harvesting.py. Keep in mind that the hindered rotor contribution to the RRHO partition functions is adapted to Gaussian output only.
Extending the supported elements of ChemTraYzer
CTY can handle the elements C, H O, N, F, S, Cl, He, Ne, and Ar by default. If you have other elements in your simulation, you need to add the maximum possible valency and the maximum possible coordination for those elements. Search for ".Val" and ".Cor" in processing.py and add the new numbers accordingly. CTY uses both values to distingush between long range interactions and actual bonds.
Python packages that are required to run ChemTraYzer:
- lammps (see https://lammps.sandia.gov/doc/Python_shlib.html)
- openbabel (version 2.4.0)
- numpy
- scipy
- sqlite3
- optional packages for plotting concentration profiles:
- pygraphviz
- matplotlib (pyplot)
Anharmonic correction to rate constants from RRHO-TST
For a short tutorial on how to correct your rate constants according to ACS Omega 2020, 5, 5, 2242-2253, please read the README.doc located in the folder small_tools/ACSOmega2020. You will need the python library TAMkin, which is not required to run ChemTraYzer.
Bug Reports:
2016-01-07: OpenBabel ignores the aromaticity of carbon atoms in SMILES in some cases
- OpenBabel adds too many implicit hydrogen atoms to carbon atoms which should be aromatic
-> analyzing.py returns a warning that the balance of elements failed, although the SMILES are balanced correctly
Bug Closed:
2015-07-30: Recrossing filter fails if final reaction of recrossing event is of type A -> A
- Reaction A -> A is removed before tracing reaction history
-> If Reaction A -> A is part of the recrossing process (only as one of the final steps), the filter tries to remove that reaction, but it cannot find it (due to previous point)
Version 1.1:
2015-07-15: (not directly related to the code) Installation of pygraphviz for windows failed
- a user reported problems with installing pygraphviz for windows 8
-> 'analyzing.py' cannot be used without pygraphviz
**SOLUTION:** pygraphviz is optional now -> 'analyzing.py' runs witout
Bug Fixes:
Version 1.1:
2015-07-08: Disturbance of atom balance in 'analyzing.py' if the initial chemical composition contains single-atom molecules.
- 'processing.py' does not recognize initializing reactions (atoms -> molecules) for single-atom molecules
- 'analyzing.py' computes initial chemical composition from initializing reactions
-> single-atom molecules are missed and concentration will become negative when these molecules are consumed. This in turn leads to wrong concentration integrals and therefore to wrong rate constants.
**SOLUTION:** Initial reactions changed so that single atoms are recognized
2015-08-31: When being initialized, Open Babel displays H and H2 molecules as empty strings
- Problems with displaying pure hydogen molecules
-> String representations of Reactions may have commas enclosing an empty string
**SOLUTION:** Identify H and H2 separately
2015-09-29: Systems without any initial bond will end with an error in the 'splitBonds' function
- Initialization of bond handler yields empty array
-> 'splitBonds' trys to access empty array and fails
**SOLUTION:** 'splitBonds' checks for empty bond array to avoid abort
2016-01-29: Recorssing filter period too long if first timestep in Bond-Order output non-Zero
- Filter period is the the product of the number of timesteps between two entries in the Bond-Order file and a user-defined number of Bond-Order steps
- the code assumes that the first timestep is zero and simply takes the second timestep as number of steps between two steps
-> the computed number of steps between two timesteps is way to large, thus the filter period is way too long and actual reactions may be filtered
**SOLUTION:** Initial timestep extraction method added
2016-01-29: Depending on the ReaxFF force field the Bond-Order between the two oxygens of a peroxide group (R-O-O.) is oscillating around 1.5
- O-O bond is oscillating between single and double bond, thus the 'open' valence of the central O is oscillating between one and zero. As a consequence, the R-O bond is periodically forming and breaking
-> the Recrossing filter is removing these oscillator events from the output until the R-O-O. undergoes further reactions (in which case R-O-O. would appear as an extremely short-lived intermediate).
**SOLUTION:** half-step bond orders allowed -> issue shifted to 0.25 BO before and after integer bond orders, which are presumably less important
Project Members:
- Enia Mudimu (admin)
- Felix Schmalz (admin)
- Florian Solbach (admin)
- Kai Leonhard (admin)
- Malte Döntgen (admin)
Discussion


Hi Felix,
I am having troubles with python3 to use this fancy tool. And it really feels painful to retrieve everything on my ubuntu 22.04 back to python2 compatible.
So, will you release a python3 version of ChemTraYzer recently?
Thanks.
Hi,
we are refactoring ChemTraYzer at the moment, making it compatible with python3 and more MD codes. We plan to release a first version this year.
Best, Felix
Hi Felix,
When I try to run CTY with -start flag (1e04), it is taking very long time to begin. Is there anyway to accelerate this time such that it will start running from the specified timestep immediately?
Thanks.
Last edit: tim rnav 2022-08-01
Hi Tim,
CTY scans through the LAMMPS/ReaxFF bond order file until the start time comes up, that can take some time. You can try deleting the part in the file from time steps 0 - 1e4 manually, and omit the start flag.
Best, Felix
Hello, I use this script to analysis the reactions in Al/C/H/O system. I have add Var 13:3, and Cor 13:6 in processing.py. I met a mistake after
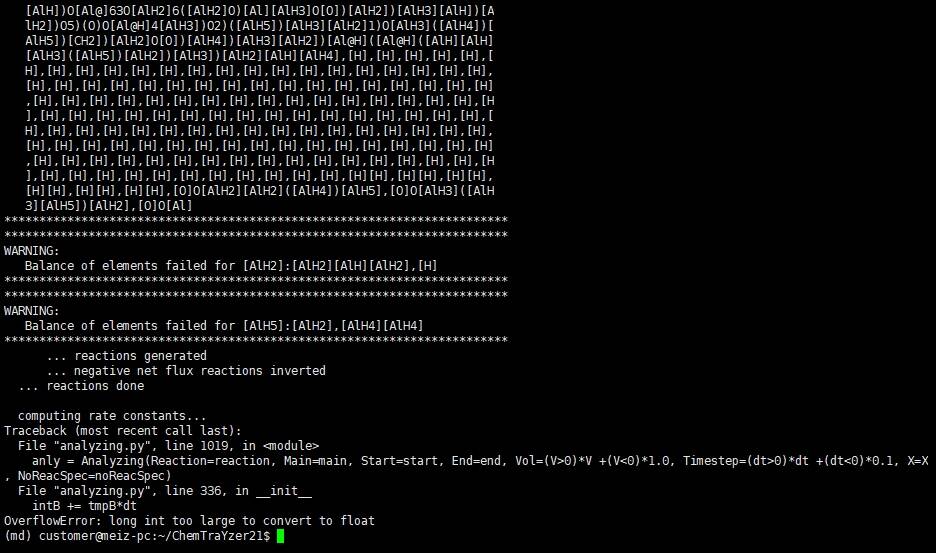
python analyzing.py 2500K.reac.reac -main reac -dp 0.1 -gen id, pointing out thatOverflowError: long int too large to convert to float. So how can I solve this problem? Thanks.Hi chengyx,
I'm guessing you are simulating a aluminum slab, which is not recommended, because the current version of CTY is developed for gas phase reactions only. How this creates the error, I can only guess:
In this step, CTY computes and sums up the reactant concentrations for a specific reaction to calculate a rate constant (see more at https://doi.org/10.1021/acs.jctc.7b00524 Eq.3). According to a test on stackoverflow, this error comes up after numbers get larger than 170 factorial.
So, there are either large numbers of reactants (tmpB) per reaction or large time intervals between the reactions (dt). In any case, CTY can't reliably handle solids.
Best, Felix
Hi, Felix,
I used CTY to simulate the reaction of Al spherical particle (D=3 nm, 700 atoms) and ethanol. As you said, it's too complex to analyze. Thereby, I downscaled the simulation system, turned the diameter of Al to 1 nm and it worked. CTY helps me to figure out the reaction mechanisms. Thank you for your reply.
Hi Felix,
The attatched file is my output from reaxff run. When I use CTY to analyzer it, my command is like,
python2 processing.py H2O2+Ag/reaxff_out.bonds -i 1:1 -i 2:8 -i 3:47 -norec 5
I already added ".Val" and ".Cor" for silver in processing.py. And I got the following error,
Processing files ...
H2O2+Ag/reaxff_out.bonds reading all steps, staring at timestep 0,
using 10000 bytes memory, a static cutoff of 0.5, considering 5
steps for recrossing and reading each 1th step of connectivity.
Identifying 1: 1, 2: 8, 3: 47
/home/xiao/Softwares/anaconda3/envs/py2/lib/python2.7/site-packages/openbabel/init.py:14: UserWarning: "import openbabel" is deprecated, instead use "from openbabel import openbabel"
warnings.warn('"import openbabel" is deprecated, instead use "from openbabel import openbabel"')
Traceback (most recent call last):
File "/home/xiao/Softwares/ChemTraYzer_2.1/processing.py", line 1210, in <module>
proc = Processing(Reader=reader, Identify=identify[f], Start=worktime[f], Storage=storage[f], Static=static[f])
File "/home/xiao/Softwares/ChemTraYzer_2.1/processing.py", line 211, in init
self.reaction, self.atom, self.molecule, self.bond[not self.switch] = self.initSystem()
File "/home/xiao/Softwares/ChemTraYzer_2.1/processing.py", line 443, in initSystem
mol[1] = self.canonical(mol[0],atoms)
File "/home/xiao/Softwares/ChemTraYzer_2.1/processing.py", line 791, in canonical
bond.SetBO(1)
AttributeError: 'OBBond' object has no attribute 'SetBO'</module>
I searched online but got no clue on how to solve this problem. Could you give a try on my out file in your computer? Thanks in advance!
Dear Zhao Xiao,
please use openbabel version 2. The warnings and errors come from version 3.x of openbabel, in which they changed some names, e.g. setBO to setBondOrder:
https://openbabel.org/dev-api/deprecated.shtml
Or you change the source code of CTY, then this might help:
https://open-babel.readthedocs.io/en/latest/UseTheLibrary/migration.html
Best, Felix
Hi Dear Developers,
I am trying to use your helpful code for processing and analyzing my bond-order results form lammps.
I could run the "processing.py" for my "Eqbonds.reaxc" file and the "reac" file was generated, However in the next step during running "analyzing.py" I get the following warning several times:
Open Babel Warning in OpenBabel::OBSmilesParser::InsertTetrahedralRef
Warning: Overwriting previously set reference id.
I could run both steps for the examples you provided, and there was no problem there.
I was wondering if any package is missing or it comes from somewhere else?
Thanks in advance!
Last edit: afsaneh khajeh 2022-08-22
Dear Afsaneh,
apparently this is an openbabel issue when reading/converting SMILES, and I don't know if they have fixed it:
https://sourceforge.net/p/openbabel/bugs/625/
It could mean that some SMILES of more complex molecules than in our example are not calculated correctly. The analyzing part computes rate constants for reactions, which is not affected by incorrect SMILES. But to be sure, check your CTY results to see if you find the molecules you expected. If two different species got the same SMILES because of that issue, those will be treated as the same.
Hope that helps,
Best, Felix
Hello,
I have two questions.
First, I get an error when I use the command 'python processing.py example/bonds.tatb -i 1:6 -i 2:1 -i 3:8 -norec 5', and the error is as follows: attempt to read from example/bonds.tatb failed. Please check whether file is broken or does not exist. File will be ignored ... I have checked the file 'bonds.tatb', and I feel nothing wrong with it. I've also tried other bonds files that I've calculated myself, and I get this error message too.
Second, is it possible to use CTY to directly analyze the xyz file already calculated by LAMMPS to obtain the reaction list?
Looking forward to the reply!
Best regards,
Xiao
Hi,
Your first question may be related to the dir name of example file. Check it carefully (examples not example). Or you can try it with your own file. If it still happens, modify everything on you linux computer to be the same version mentioned on the website.
As for your second question, I dont think it can be done without pre-processing. You can also ask the developers.
Good luck!
Hello,
Thank you very much for your reply!
The previous problems have been solved. Now I have a new problem and would like to ask you for some advice. The file with the .reac extension obtained from the processing.py file has information about reaction species. For example, there are species with ":" symbols such as "O=O:C[C]", and species with "C=C1OO1", what do they mean?
Thanks again for your reply!
Best regards,
Xiao
Hello Xiao,
ChemTraYzer relies on connectivity changes in the trajectory. So, it's not enough to just input XYZ. However, we are working on a new version which will include a basic bond guessing algorithm for exactly this case.
The colon ":" is our choice of separating SMILES from one another, because this character doesn't appear in the SMILES generated by openbabel. So, "O=O:C[C]" are two species, and "C=C1OO1" is one (e.g. see https://molview.org/?q=C=C1OO1).
Hope that helps,
Kind regards,
Felix
Hi Felix,
When I use CTY to analyzer it, my command is like,
python2 analyzing.py bonds5-1200.reaxc.reac -main bonds5-1200 -p CCCCCCCC/C=C/CCC[CH]CC[O]:C=O,CCCCCCCCC=CCCCC=C
error:
Traceback (most recent call last):
File "analyzing.py", line 1030, in <module>
if plot: anly.drawProfile(Plot=plot, Timestep=timestep)
File "analyzing.py", line 484, in drawProfile
ax.append(plt.subplot2grid((nsub,msub),(i%nsub,i//nsub),colspan=1,rowspan=1))
File "/home/zxs/.local/lib/python2.7/site-packages/matplotlib/pyplot.py", line 1237, in subplot2grid
colspan=colspan)
File "/home/zxs/.local/lib/python2.7/site-packages/matplotlib/gridspec.py", line 63, in new_subplotspec
subplotspec = self[loc1:loc1+rowspan, loc2:loc2+colspan]
File "/home/zxs/.local/lib/python2.7/site-packages/matplotlib/gridspec.py", line 161, in getitem
[_normalize(k1, nrows), _normalize(k2, ncols)], (nrows, ncols))
File "/home/zxs/.local/lib/python2.7/site-packages/matplotlib/gridspec.py", line 153, in _normalize
raise IndexError("invalid index")
IndexError: invalid index
Please give me some suggestions!Thanks!
Best regards,
Biao</module>
Hi Felix,
I have noticed that the event counts of few reactions in the "reac.reac.tab" file shows "-1". So what does the negative value mean? Is there a problem with my input file?
Hi Jingyi Yu,
it just means that the reverse reaction occured. Reverse in regard to what is printed in the third and fourth line.
Best, Felix
Hello
I have some trouble about processpy.
... list of species indices generated
... list of reaction indices generated
Traceback (most recent call last):
File "analyzing.py", line 1012, in <module>
anly.depictMolecules(Resize=resize)
File "analyzing.py", line 644, in depictMolecules
call(['mkdir',folder])
File "C:\Python27\lib\subprocess.py", line 172, in call
return Popen(popenargs, *kwargs).wait()
File "C:\Python27\lib\subprocess.py", line 394, in init
errread, errwrite)
File "C:\Python27\lib\subprocess.py", line 644, in _execute_child
startupinfo)
WindowsError: [Error 2]
please help me!
Bestwish</module>
Hi Gaohang,
The error seems to come up while creating a folder. What name did you choose for your files (the "-main" option)? The folder will contain this name and some characters are not allowed (e.g. "<>\ / |"). If that seems correct, write permissions might be the problem here.
Best, Felix
Hi Felix,
Thank you for your help. I deleted the call command in the code. Although there are some problems with the running interface, the result looks good. Now I have another problem. I run the process module to process my file, but it has an error. The error code is as follows: Processing files
bonds.reaxff reading all steps, staring at timestep 0, using 10000
bytes memory, a static cutoff of 0.5, considering 100 steps for
recrossing and reading each 1th step of connectivity.
Identifying 1: 8, 2: 14, 3: 9, 4: 1
Traceback (most recent call last):
File "processing.py", line 1215, in <module>
timestep, tmp = proc.processStep(Recrossing=recrossing, Steps=recsteps, Skip=skip, Close=True)
File "processing.py", line 279, in processStep
self.timestep, self.bond[self.switch] = self.extractBonds(Step=self.step, Bond=self.bond[self.switch], Static=self.static)
File "processing.py", line 570, in extractBonds
self.atom[ID].q = Q
KeyError: 226
I am considering whether chemtrayzer cannot handle the increase of particle number in the system, because I adopted the deposition command in lammps. The number of particles in the system in step 0 is 225, and the number of particles in the system in step 50 is 227. I don't know if my guess is correct. I hope you can help me.
Best, Gaohang</module>
Last edit: gaohang 2023-02-28
Hello Felix Scgmalz,
Thank you for providing such useful software.
I have encountered the following problems while using it. I am using ChemTraYzer_2.0 version. My research system includes elemental fluorine. In the data file, #1 is carbon, #2 is oxygen, #3 is fluorine, and #4 is hydrogen. The error is reported as follows.
ERROR:
some elements are not supported by the internal valence list.
Please extend the list of supported elements by adding to
Processing.Val
I'm not quite sure how to add the elements, can you provide some guidance?
Best wish to you
Thank you for contacting us and sharing your interesting application.
If your version corresponds to 2.0 from the sourceforge download site, the issue should be in the “processing.py” file.
Therein, lines 154 and 155 contain dictionaries that assign to element numbers the corresponding valencies. In the version from sourceforge, these do contain values for element number 9 (flourine):
self.Val = {1: 1, 2: 0, 6: 4, 7: 5, 8: 2, 9: 1, 10: 0, 16: 6, 17: 1, 18: 0}
self.Cor = {1: 1, 2: 0, 6: 4, 7: 3, 8: 2, 9: 1, 10: 0, 16: 6, 17: 1, 18: 0}
Is this also the same for your version?
Initialization of a “Processing” object can take an “Identify” dictionary. This may contain your assignment of enumeration to element numbers. What do you give as “Identify” dictionary? According to your information, I guess it should look like (1:6, 2: 8, 3: 9, 4:1)
Do you use LAMMPS? Which force field and parametrization do you employ?
Best Regards
Wassja A. Kopp
On behalf of the ChemTraYzer Team
Thank you for your reply.
With your help, the problem we encountered has been solved. Thank you for providing such an interesting package!
Dear Felix,
Thank you for providing us with such useful software!
The "add element" problem we encountered before has been solved.
I have three questions for you.
1) I don't quite understand the statement "python processing.py bonds.tatb -n 0 -i 1:6 -i 2:1 -i 3:8 -norec 100"
-n I don't quite understand, the readme file explains "ALL timesteps which have been written to 'bonds.tatb"
Does this timestep refer to the timestep recorded in the bond.tatb file or the timestep set by the in.reax file,(0.1fs)
Here are the first few lines of my bond.tatb file.
"# Timestep 7
Number of particles 1670
Max number of bonds per atom 4 with coarse bond order cutoff 0.300
Particle connection table and bond orders
id type nb id_1... .id_nb mol bo_1... .bo_nb abo .bo_nb abo nlp q"
2)"-norec xx"I can't understand how to set this parameter? Will setting the parameter have an effect on the calculation results?
3) The first line of the "reac.rate" file: "R<id>;S<id>'s;Formula's;SMILES;k;klo;kup;N"
Does k represent the rate constant? And what does klo;kup;N stand for?
Best wish to you</id></id>